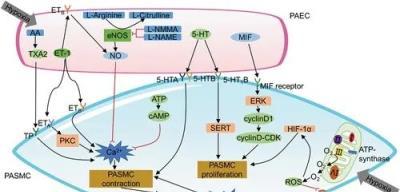
肉雞腹水綜合徵的發病機制
肉雞AS的發生涉及複雜的多因素過程,其發病機制尚不完全清楚。缺氧、絲裂原、內皮源性血管活性分子、氧化應激、炎症因子、離子通道、受體、神經遞質、非表觀遺傳現象和轉錄因子介導均參與肉雞AS的調節(圖1 )。
圖 1.肉雞腹水綜合徵的發病機制。
缺氧在肉雞腹水綜合徵中的作用
研究表明,代謝性缺氧是引起缺氧性PAH的關鍵原因。缺氧可導致一系列病理生理變化,包括外周血管舒張、右心室肥厚、心輸出量增加等(Wideman等,2015) 。引文2000)。麥克斯韋等人。報道稱,快速生長的肉雞發生低氧血癥與通風不良或環境缺氧無關。事實上,引起低氧血癥的主要原因是肺血管容積不足以容納迅速增加的心排血量,使肺血流速度過快,無法有效交換氣體,從而引起低氧血癥(Maxwell等,2017 )。引文1990年;洛倫佐尼、安東尼和維德曼引文2008)。肺血管容量不足主要是由肺血管收縮和肺血管重塑(PVR)引起的。
低氧血癥和肺動脈高壓之間的關係很複雜。理論上,低氧血癥可通過降低全身血管阻力(缺氧性全身血管舒張)或增加肺血管阻力(缺氧性肺血管收縮)而導致肺動脈高壓(Wideman 等,2017)。引文2000;魯伊斯-費里亞和維德曼引文2001)。血管收縮主要是由於血管活性因子失衡,即血管收縮介質過多或血管舒張介質缺乏所致。遠端肺微動脈不可逆重塑的形成也是肺動脈高壓的主要病理特徵,稱為叢狀病變(PLS)。PVR是指肺血管壁細胞和細胞外基質由於缺氧、高血流剪下應力和炎症等損傷因素的影響而發生血管結構的改變。引文2018年)。PVR的主要特徵是肺小動脈內側平滑肌增生和非肌性小動脈肌化。血管重塑包括內皮細胞損傷、平滑肌細胞增殖、外膜纖維增殖等解剖學變化。表現為血管各層增厚,其中以中膜增厚最為顯著。重建後肺血管壁增厚,管腔縮小,血流阻力增加,血管順應性下降,肺迴圈對血流量增加的適應性大大降低,容易引起肺動脈高壓。 。肉雞肺血管形態的改變進一步增加了肺血管的阻力,在AS的發生發展中發揮了重要作用。一般認為,PVR是對肺動脈壓升高的適應性反應,是肉雞形成穩定AS的基礎(Wideman和Hamal)引文2011年;維德曼等人。引文2011年,引文2015)。
氧感測能力是細胞為適應長期進化而形成的重要生理功能,哺乳動物細胞對缺氧的適應性調節是通過改變一系列基因表達來實現的。缺氧誘導因子-1 (HIF-1) 是調節這些基因表達的最重要的轉錄因子之一。它主要由兩個亞基HIF-1α和HIF-1β組成。其中,HIF-1β在常氧和缺氧條件下均可以組成型表達。然而,HIF-1α僅在缺氧情況下在細胞核中表達,是決定HIF-1活性的功能亞基。HIF-1α被缺氧訊號啟用後,與HIF-1β結合形成穩定的HIF-1。Hif-1的穩定表達可調節數十種缺氧相關基因的表達,如促紅細胞生成素(EPO)、糖酵解酶、血管內皮生長因子 (VEGF) 和 ET-1。研究證實,這些靶基因大多參與哺乳動物肺動脈高壓的形成(Shimoda、Yun和Sikka)引文2019)。HIF-1α基因與哺乳動物肺動脈高度在實驗動物和人類醫學中的研究成果為探索肉雞腹水的形成機制提供了非常重要的思路。通過雞胚心室肌細胞HIF-1α基因的cDNA克隆,獲得HIF-1α基因的全長cDNA序列。測試發現雞HIF-1α的氨基酸序列與人HIF-1α的氨基酸序列有79%的同源性,表明HIF-1α基因在進化上高度保守(Takahashi等,2017)。引文2001)。此外,在過量鹽誘導的AS肉雞的心臟和肺中發現,隨著肺動脈壓的升高,過量鹽組AS肉雞中HIF-1αmRNA的表達量逐漸增加(Zhang等,2017)。引文2013)。這些發現表明 HIF-1α 可能參與 AS 的發生。曾等人。發現肉雞肺內HIF-1α、VEGF和血管內皮生長因子受體2(VEGFR2)的mRNA水平呈正相關。VEGF mRNA 的表達與肺血管壁面積與總血管面積的比值(WA/TA)和平均肺小動脈中層厚度(mMTPA)呈顯著正相關。
氧化應激對AS的影響
氧在體內主要參與線粒體呼吸和氧化磷酸化生成ATP,其最終產物主要是水。但氧代謝過程中也會產生一系列中間產物,包括超氧陰離子(O 2 - )、羥自由基、氫過氧自由基、H 2 O 2等氧自由基。、單線態氧、三線態氧等。這些物質具有很強的氧化能力,統稱為ROS。在缺氧條件下,線粒體發生裂變併產生離散的、小尺寸的圓形線粒體。此後,失敗的線粒體要麼被線粒體自噬過程吞噬,要麼這些受損線粒體積累的增加導致 ROS 進一步惡化(Fuhrmann 和 Brune)引文2017)。
臨床發現,與健康人PASMCs相比,PAH患者PASMCs的膜電位和mROS含量降低(Bonnet等,2017)。引文2007)。PASMC 在慢性缺氧和野百合鹼誘導的動物 PAH 模型中也表現出相同的現象(McMurtry等,2017)。引文2004)。這些研究表明 PAH 的線粒體代謝功能失調。在此基礎上的進一步研究表明,mROS的減少會抑制細胞的氧化還原狀態,從而形成假缺氧狀態(Marsboom等,2015)。引文2012)。近年來許多研究表明,氧自由基介導的機制可能是肉雞在假性缺氧狀態下發生AS的主要原因。關於自由基在肉雞AS形成中的作用以及自由基的清除,目前已有大量的實驗研究。對 AS 肉雞抗氧化狀態的研究表明,自由基可能在肉雞 AS 的發病機制中發揮關鍵作用(Enkvetchakul等,2017)。引文1993)。Maxwell等研究認為心肌中乳酸脫氫酶(LDH)和細胞色素氧化酶的增加、線粒體中Ca 2 的沉積以及超氧化物的增加都是右心室心肌損傷的結果。利用缺氧成功誘導肉雞AS,發現AS肉雞心肌線粒體上存在大量H 2 O 2 ,而陰性對照組則很少或沒有H 2 O 2 (Maxwell 、羅伯遜和法誇森引文1996)。肉雞發生腹水後,心肌細胞產生大量過氧化氫,導致心肌損傷。研究發現AS雞心肌和胸肌線粒體呼吸過程中發生電子洩漏。表明肉雞細胞對氧的利用率降低,過氧化氫和ROS含量增加,肉雞腹水發生率增加(唐等,2017)。引文2002)。此外,有研究觀察到低溫條件下肉雞體內和體外培養的血管內皮細胞中丙二醛(MDA)和ROS的含量增加(Arab等,2015)。引文2006年;潘等人。引文2007)。
炎症影響肉雞腹水綜合徵的形成
炎症是肺動脈高壓發病機制中的一個新興概念,大量研究表明炎症與肺動脈血管重塑密切相關(Berghausen等,2017)。引文2019)。越來越多的證據表明血管周圍炎症在 PAH 和 PVR 的發生和進展中發揮作用。PAH 患者體內可檢測到大量細胞因子、趨化因子和炎症介質在 PAH 肺血管周圍積聚(Stacher等,2017)。引文2012)。同時,血管和內皮細胞、平滑肌細胞和成纖維細胞可以改變其表型,影響PAH的形成。在一項針對 AS 肉雞的研究中,發現炎症細胞標記物在肉雞 CVL 中強烈表達,並且在血管周圍區域有由 T 淋巴細胞、B 淋巴細胞和巨噬細胞組成的浸潤物 (Hamal 等,2017)。引文2012年;克魯斯等人。引文2012)。丹蔘酮IIA(TIIA)是一種抗炎、降壓藥物,當AS肉雞飼餵新增TIIA的日糧時,發現IL-6、IL-1β、NF-κB和p38蛋白等炎性細胞因子的濃度降低在 AS 肉雞中。這些發現表明,炎症因子參與肉雞 AS 的發病機制,而丹蔘酮 IIA 可以保護肉雞免受 PAH 的侵害(Hu等,2017)。引文2017)。進一步研究了肉雞AS與炎症因子的關係。研究發現,AS肉雞肺動脈中巨噬細胞遷移抑制因子(MIF)的增加,通過細胞外訊號調節激酶(ERK)訊號通路上調cyclinD1,誘導肺血管平滑肌細胞增殖,引起肺動脈粥樣硬化。重塑和高血壓(Li et al .引文2017)。這些研究表明,免疫細胞和炎症細胞因子可能在 CVL 的發生過程中發揮關鍵作用,並且它們可能通過與肉雞肺部血管增殖分子和生長因子相互作用來促進 PLS 的發生。
結論
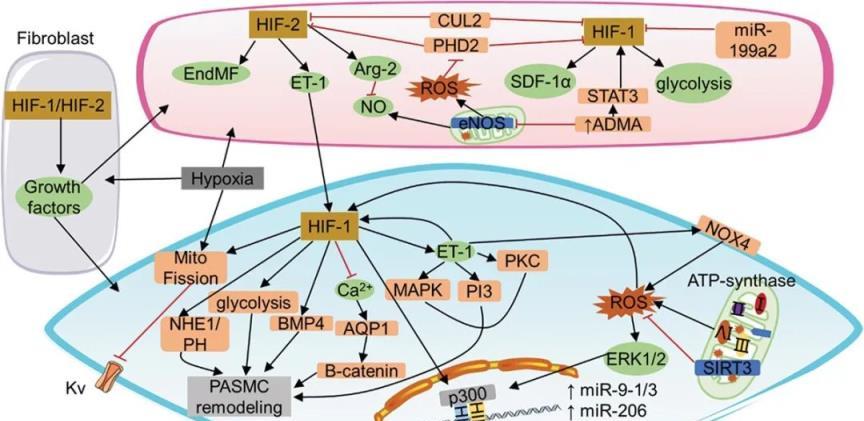
肺動脈高壓的發病機制是由血管收縮、微血栓形成和小肺動脈重塑共同驅動的。人類和哺乳動物的大量文獻證實,血管活性因子、ROS、缺氧誘導因子、炎症因子和表觀遺傳現象的調節可能在可能導致肺動脈高壓的機制的發病機制中發揮重要作用(圖2、圖3 ))。所有這些因素都存在於大多數導致肺壓升高的實體中,儘管持續時間在不同程度上對病程有影響。在肉雞AS發病機制的探索中,發現大量肺動脈高壓調節通路與人類PAH相同。因此,探討肉雞AS發病機制也可為人類PAH的防治提供參考。其中,免疫系統在哺乳動物PAH中的基礎作用越來越受到重視。炎症、代謝過程甚至腫瘤發生樣變化已被認為是 PAH 發病機制的關鍵範例。在肉雞中,免疫系統和炎症對肉雞 AS 發病機制的影響很少研究。進一步探討炎症因子對肉雞AS發病機制的特異性調控可能有助於更深入地瞭解肺動脈高壓的發病機制。它不僅為肉雞AS的潛在發病機制提供了更深入的瞭解,而且為人類肺動脈高壓的治療提供了理論支援。
圖 2.肺動脈高壓的血管收縮機制
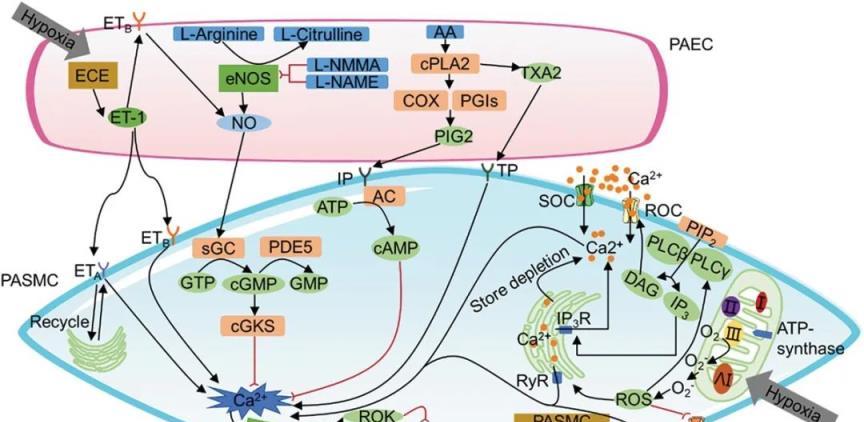
圖3。肺血管重構的機制
來源:雞保姆,作者:默罕默德 沙汗等